
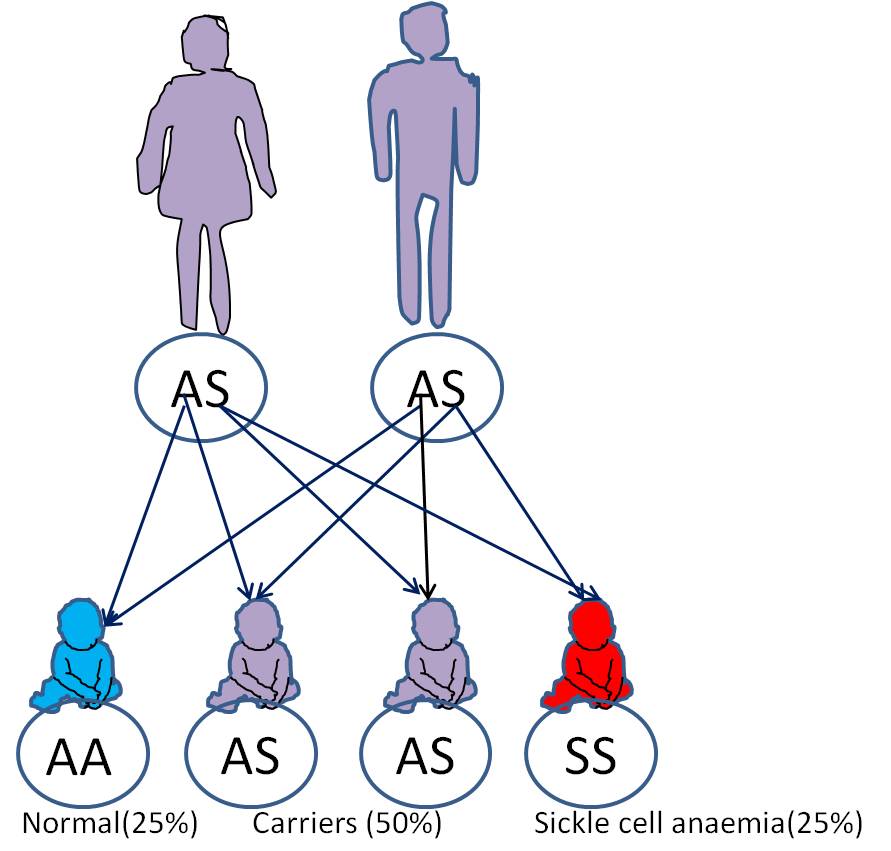
Sickle cell anemia is one of the common causes of anemia. Anemia is defined as a state of reduced red blood cells or the hemoglobin which they contain. Sickle cell anemia is a genetic disorder passed on from parents to offsprings in an autosomal recessive pattern. This means that its expression does not depend on the gender of the offspring (autosomal) and the offspring needs to inherit one abnormal gene each from both parents in order to have the disorder (recessive). There are however other conditions which may arise if an offspring inherits the abnormal gene from only one parent. These, along with the SCA are collectively grouped as Sickle Cell Disease, SCD. These offsprings with only one defective gene are referred to as carriers of the Sickle cell trait because they can also pass it on to their children and can have children with sickle cell anemia if the other parent is also a carrier of the trait. Carriers usually appear normal, without any form of anemia but some may have minor anemia and other symptoms experienced by SCA patients.
The genetic abnormality in Sickle cell disease is a mutation in a single nucleotide of the gene coding for β globin, located on chromosome 11. Hemoglobin, a protein found in red blood cells, is normally made up of 2α and 2βglobin chains. Red blood cells are able to carry oxygen around the body mainly because of the presence of hemoglobin in them. Most of the oxygen in blood is chemically bound to hemoglobin and release when needed. Normal hemoglobin is called hemoglobin A (Hb A). The gene mutation in SCD gives rise to the production of structurally defective β globins which in turn cause the formation of abnormal hemoglobin molecules called hemoglobin S (Hb S). Under conditions of low oxygen concentration, the deoxygenated Hb S molecules aggregate, forming fibrous polymers. This does not occur with normal hemoglobin. The fibrous precipitates formed cause the red blood cell to change its shape from the normal disc shape to a sickle shape. Sickle shaped cells are not elastic and are therefore easily destroyed as they pass through small blood vessels and spleen. This gives rise to the chronic anemia and jaundice (yellow sclera of the eyes) seen in SCA. Sickle cells have a lifespan of about 10-20 days compared to 120 days for normal cells. This rate of destruction is too fast to be compensated by production of new cells. Also, many sickle cells can aggregate and block small vessels giving rise to the various forms of vaso-occlusive crisis and other complications. The symptoms however do not
occur until after the age of 6 months when the level of the fetal hemoglobin, Hb F, decreases and production of Hb S increases.
occur until after the age of 6 months when the level of the fetal hemoglobin, Hb F, decreases and production of Hb S increases.

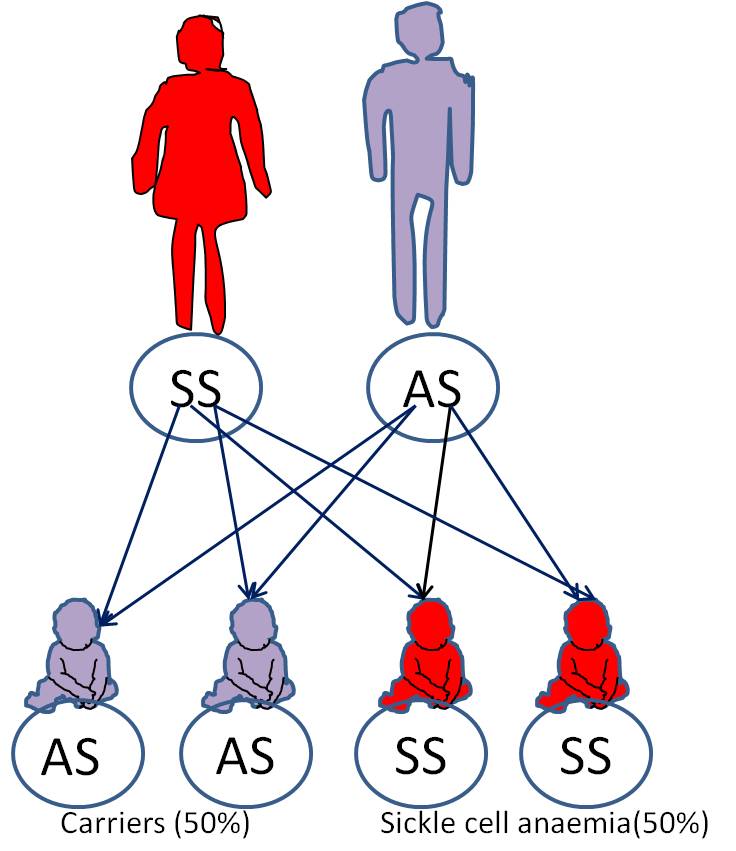
Whereas if one parent is a carrier and the other has Sickle cell anemia, there is, for each pregnancy, a 50% chance of the child being a carrier and 50% chance of the child having SCA.
Again, if one parent is completely free of the defective gene and the other is a carrier, there is, for each pregnancy, a 50% chance of the child being normal and 50% chance of being a carrier.

This is how the gene has been propagated from generation to generation and from one region to another.
People who are homozygous for the sickle cell gene i.e. those with sickle cell anemia usually have some common features which give them a unique look. This is called the “sickle cell habitus”. These features include thin, lanky stature with long legs, frontal bossing (prominent forehead), pallor, jaundice, long thin fingers.
Complications of SCA include the following:
- Gallstones and cholecystitis,
- Overwhelming infection (due to destruction of spleen)
- Decreased immune reactions due to hyposplenism (malfunctioning of the spleen).
- Stroke
- Hand and foot syndrome
- Avascular necrosis of the hip and other major joints,
- Priapism (painful, undue, long lasting erection) and damage to the penis.
- Acute papillary necrosis in the kidneys.
- Leg ulcers.
- Osteomyelitis (bacterial bone infection)
- Addiction to opiate analgesics
- Blindness.
- spontaneous abortion,
- Pulmonary hypertension
- heart failure
- Chronic renal failure
Early diagnosis is critical in sickle cell disease management. Diagnosis is by hemoglobin electrophoresis.
Treatment involves:
Treatment involves:
· Prevention of severe anemia; by regular intake of folic acid tablets. (Never take iron tablets because there’s already excess iron released into the blood from excess hemolysis)
· Blood transfusion to treat severe anemia and to dilute the hemoglobin S with normal hemoglobin
· Drinking about 10 glasses of water per day to prevent dehydration which precipitates pain crisis or to treat it if already occurring
· Analgesics for pain
· Prompt treatment of infections
- Hydroxyurea
- Bone marrow transplant
sickle cell anemia.
No comments:
Post a Comment